Introduction To Pharmacokinetics
Pharmacokinetics sometimes also abridged as PK, is a pharmacology branch devoted to the determination of the substances fate controlled externally to a living organism. The interested substances comprise hormones, pharmaceutical agents, toxins and nutrients. The term pharmacokinetics is the study in relation to movement of drugs in the human body. It includes four main pathways, such as absorption, distribution, metabolism as well as elimination (PHARMACOKINETICS OF ASPIRIN, 2011). If a drug is taken orally with a liquid, tablet, capsule or suspension then more factors required to be considered like the rate of dissolution and the disintegration of drug. If a drug is obtained with an IV bolus or an IV infusion, the procedure starts at the pathway of absorption. Thus, the present report is prepared to comprehend how a solid drug tablet like aspirin moves in the body of human. In order to better understand the whole process, researcher will demonstrate each pathway under separate headings providing good presentation through appropriate diagrams (Aspirin: Pharmacokinetics, 2012).
Overview of Aspirin
Aspirin, which is also known as ASA or acetylsalicylic acid, is the salicylate ester of acetic acid. The compound takes place as a white, crystalline powder or needle-like or tabular crystals. It is fragile acid with a 3.5 pka. Aspirin is little soluble in water and generously soluble in alcohol. Every gram of aspirin includes more or less 760 mg of salicylate. Aspirin is often used as analgesic (to reduce minor pains and aches), as an antipyretic (to lessen fever) and as an anti-inflammatory medicine. Aspirin was initially isolated in 1897 by Felix Hoffmann, a chemist of the German organization, Bayer (Pediatrics, 2009).
If your dream is to get top grades, get a rewarding assignment service from us.
Brilliant Assignment ServicesToll Free: +61 482070482
help@instantassignmenthelp.com.auSalicylic acid is the key metabolite of aspirin which is an integral part of animal and human metabolism; whereas in latter to a large extent, it is attributable to diet and a considerable part is synthesized endogenously. The main side effect of aspirin is stomach bleeding, gastrointestinal ulcer and tinnitus, particularly in higher doses (Graham and et. al., 2004). In adolescents and children, aspirin is no longer signified to control symptoms of chickenpox, viral illness or flu, due to the risk of Reye’s syndrome. Adult’s aspirin medicines are manufactured in standardized sizes that vary a little from one country to another, for instance, 325 mg in the USA and 300 mg in Britain. Smaller doses are founds on these standards; like 75- or 81-mg medicines; there is no medicinal implication in the minor variation (Ecotrin (Aspirin Enteric Coated) - Description and Clinical Pharmacology, 2012).
In general, doses are taken four times in a day for arthritis or fever, in the case of adults. For the reason to prevent myocardial infarction in an individual with suspected or documented coronary artery disease, much lower doses are taken at one time on daily basis (Demir, 2011).
ADME Scheme
Pharmacokinetics is separated into varied areas including the rate plus extent of absorption, distribution, metabolism along with excretion which is also called as ADME scheme:
- Absorption – It is the procedure of a substance that enters in blood circulation (McKay and et. al., 2011).
- Distribution – It refers to dissemination or dispersion of substances all through the tissues plus fluids of the body.
- Metabolism or Biotransformation – This is the irreversible transformation of parent compounds into metabolites of daughter (Rainsford, 2004).
- Excretion – It is the process of removing substances from the body. Rarely, some drugs irrevocably accumulate in the tissue of body.
Elimination is the outcome of excretion and metabolism. Therefore, pharmacokinetics defines that after administration, how the body influences a particular drug (Burton, 2006). Pharmacokinetics properties of drugs might be affected by factors such as the dose of administered drug and site of administration. These might affect the rate of absorption. A fifth procedure, i.e. Liberation has been highlighted as playing a significant role in pharmacokinetics. Liberation is the method of discharging drug from the formulation. Hence, LADME can at times be used in position of ADME in reference to the central part of pharmacokinetics (Tozer and Rowland, 2006).
Outline of Aspirin drug pathways
The below given image depicts a concise outline of distinct pathways taken on its route by a drug tablet through the body of human entity.
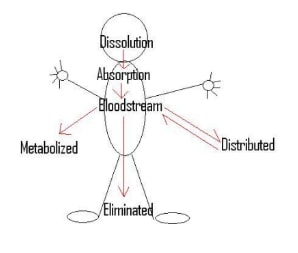
Dissolution
Once a solid medicine is ingested, it passes through the track of GI (Gastrointestinal) where firstly it is disintegrated and then dissolved. The rate, which it takes for dissolving a tablet, is referred as dissolution of drug. This procedure can be represented by the equation of Noyes-Whitney (Lewis, 2000). As shown in the diagram given below, the drug tablet is primarily concentrated to granules and then to small elements all the way through disintegration. At some point in all these three phases, dissolution takes place. The more the body breaks down the tablet, the better rate of dissolution it produces. Since, the body collapses medicine, some of the particles gain negative or positive charges, which makes them icons. Such icons are excreted all through the human body or taken to other location for additional use (Tran and et. al., 2007). The icons will not disseminate across the membrane as well as into the bloodstream. Moreover, if there is any food or another drugs exist in the stomach, then it could impede with the rate on which table is fragmented together with the dissolution rate. Also, the absorption of the drug will also be reduced.
Increase Your Odds of Success With Our
- Scholastic academic documents
- Pocket friendly prices
- Assured reliability, authenticity & excellence
Assumptions
- Dissolution only takes place in final phase in case of small particles.
- Entire dissolution.
- None of the particles have a negative or positive charge.
- No presence of food in stomach.
- Complete medicine dissolves (Demir, 2011).
Absorption
While a drug is being engrossed into the bloodstream, it could be transported by three ways across a membrane. First one is active transport, which needs an energy input, ATP, through the cell. It facilitates the medicine to go from a less absorption to high absorption. Second is an easy diffusion, allowing only small and uncharged molecules to transport across membrane. Finally, the facilitated diffusion which needs a carrier protein and will allow uncharged, large molecules or charged plus small molecules to transport across the membrane. Both facilitated plus small diffusion move the aspirin from a part of high to low absorption in which energy is not required. In general, instant release aspirin is completely and well absorbed from the GI track (Rainsford, 2004). The absorption rate from the GI track is based on the form of dosage, the absence or presence of food, physiological factors and gastric pH (the nonexistence or existence of buffering agents or GI antacids).
Assumptions
- Every medicine absorbed into bloodstream.
- No charged molecules.
- Assume stable condition diffusion.
- Aspirin is transported through passive diffusion.
- Regular thickness of membrane (Tozer and Rowland, 2006).
- No prior medicine existing in the bloodstream.
- The aspirin travels from high to low absorption.
Distribution
The key system of transferring nutrients to the organs and tissues of human body is via the bloodstream. When the nutrients appear at a cell which is deficient, it will develop a concentration gradient within the blood vessel and cell. Then, the nutrients will seek to disperse from the high intense blood to low intense cell providing the adequate nutrients to the body’s cells (Tran and et. al., 2007).
When a drug is introduced into the bloodstream of body, then its distribution rate is dependent upon numerous aspects. Firstly, the tissues with maximum blood flow, obtain the drug first. Secondly, if the drug is connected to a plasma protein, it could just go anywhere the proteins go. Third, the medicine could enter tissues, which are very lipid soluble. Finally, distribution is dependent on the drugs ionization degree and unionized drugs could anywhere in the human body. These four aspects also determine where the drug will finish up in the body (Lewis, 2000).
In case of aspirin, salicylic acid is greatly dispersed to all fluids and tissues in the body including the fetal tissues, central nervous system and breast milk. The maximum concentrations are found in the liver, plasma, lungs, heart and renal cortex. The salicylate protein binding is concentration-dependent, such as non-linear. At less concentration (< 100 mcg/mL) around 90% of plasma salicylate is bound to albumin, whereas at higher concentrations (> 400 mcg/mL) only around 75% is bound. The early indicators of salicylic overdose together with tinnitus, i.e. ringing in the ears, takes place at plasma concentrations about 200 mcg/mL. Severe poisonous effects are connected with levels > 400 mcg/mL (Mason, Freedman and Jacobs, 2004). Here, the diagram is given, which shows in what way drug moves into the body via going through with multiple paths:
Assumptions
- Assume a stable heart beat.
- The drug is evenly distributed all through the body.
- No earlier drugs available in the body.
- No charged particles.
- No ionized particles (Paterson and Lawrence, 2001).
Metabolism
The aim of metabolism is to make it simple for the drug to execute from the human body. The main site for metabolism is the liver and drugs are metabolized by reduction, oxidation, conjugation, isomerization or condensation. Most of the drugs are metabolize in two segments. The first stage includes cutting functional groups by reduction, hydrolysis or oxidation. The second phase consists of conjugation by an endogenous material. In the trice phase of metabolism, polar metabolites are developed and are rapidly excreted with the kidneys in the urine form. Those metabolites, which are created in first phase of metabolism, are not readily excreted (Kubitza and et.al., 2006).
Aspirin is quickly hydrolyzed in the plasma to salicylic acid, as plasma intensities of aspirin are significantly undetectable 1 to 2 hours later than dosing. Salicylic acid is initially conjugated in the liver, in order to form salicyluric acid, an acyl glucuronide, a phenolic glucuronide as well as numerous minor metabolites. Salicylic acid has plasma of half life, i.e. around 6 hours. Salicylate metabolism is saturable plus entire body clearance lessens at higher serum concentrations because of limited capability of liver to shape both phenolic glucuronide and salicyluric acid. By following noxious doses (10-20 grams (g)), the half life of plasma might be increase to over 20 hours (Levy, 2012).
Assumptions
- All drugs are metabolized.
- No another drugs are available in the liver.
We believe in serving our customers with the most reliable assignment help
Elimination
Once a drug has been utilized to its complete potential, it is excreted from the human body. The drug elimination task follows five processes, such as hydrolysis, conjugation, reduction, oxidation and lastly excretion. Drugs could be excreted by salivary glands, urine, lungs in gas form, tears, sweat, fecal matter and milk (Seymour and Rawlins, 2012).
In aspirin, elimination process follows zero order pharmacokinetics; i.e. the rate of medicine elimination is invariable with regards to plasma concentration. Renal excretion of unaffected drug based upon urine pH. As urinary pH grows above 6.5, the renal authorization of free salicylate rises from 5 percent to 80 percent. In the management of salicylate overdose, the key concept of urine is Alkalinization. By following doses of therapeutic, around 10 percent excretion is found in urine as salicylic acid, 10 percent phenolic, 5 percent acyl glucoronides of salicylic acid and 75 percent as salicyluric acid (Husted and et.al., 2006).
Conclusion
The pharmacokinetics of Aspirin starts as soon as the medicine is ingested by human body. In general, instant release aspirin is completely absorbed and dispersed to all fluids and tissues in the body. These are inclusive of fetal tissues, central nervous system and breast milk. This is followed by its quick hydrolysis to salicylic acid. The elimination process also follows a zero order pharmacokinetics.
References
- Levy, G., 2012. Clinical pharmacokinetics of salicylates: a re-assessment. British Journal of Clinical Pharmacology. 10 (2). pp.285-290.
- Lewis, C., 2000. Food and medicine incompatibility. Nutrition and Food Science Journal. 91 (5). pp.112-117.
- Mason, J. P., Freedman, E. J. and Jacobs, K. A., 2004. Aspirin resistance: current concepts. PubMed. 5 (3). pp.156-63.
- Paterson, R. J. and Lawrence, R. J., 2001. Salicylic acid: a link between aspirin, diet and the prevention of colorectal cancer. QJM: An International Journal of Medicine. 94 (8). pp.445-448.
- Seymour, A. R. and Rawlins, D. M., 2012. Efficacy and pharmacokinetics of aspirin in post-operative dental pain. British Journal of Clinical Pharmacology. 13 (6). pp.807-810.
- Tran, E., and et.al., 2007. Vitamins and minerals: powerful medicine or potent toxins. Journal of Food Science. 37 (2). pp.67-73.

UPTO50%
Avail The Benefit Today!
To View this & another 50000+ free









